
News
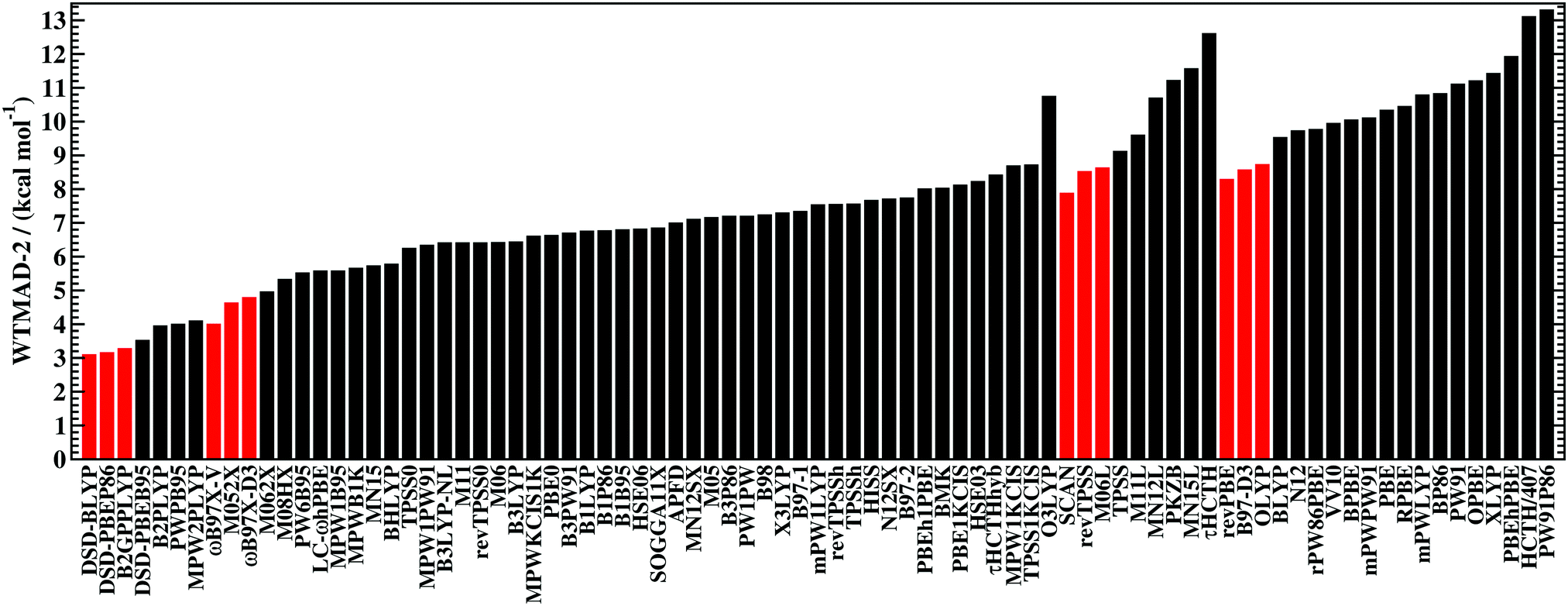
Goerigk et al. recently published the GMTKN55 database for general main group thermochemistry, kinetics, and noncovalent interactions, containing 1505 relative energies based on 2462 single-point calculations. Based on the weighted total mean absolute deviation (WTMAD-2) metric (see below), ωB97X-V is the best Rung 1-4 functional tested, with a WTMAD-2 of 3.98 kcal/mol. The next best local or hybrid functional is M05-2X-D3(0), coming in at 4.61 kcal/mol (more than 15% worse). Across all non-covalent interactions in GMTKN55, ωB97X-V achieves a WTMAD-2 of only 3.32 kcal/mol, outperforming all tested functionals, including double hybrids. The next best performer is DSD-BLYP-D3(BJ), with a WTMAD-2 of 3.55 kcal/mol, while the next best local or hybrid functional is BHLYP-D3(BJ) with a WTMAD-2 of 4.66 kcal/mol. Thus, compared to models in and below its class, ωB97X-V represents a 15% improvement overall, and a 30% improvement for non-covalent interactions. It will be interesting to see the performance of ωB97M-V and B97M-V on this dataset.
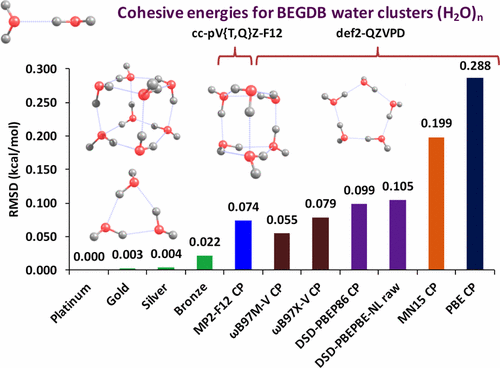
Recently, Manna et al. revised the binding energies of the 38 water clusters (dimers through decamers) in the BEGDB water cluster dataset. Using the updated reference values, a series of density functionals and wave function methods were benchmarked. The results indicate that ωB97M-V is the best tested method, with a cohesive energy RMSD of only 0.055 kcal/mol, outperforming MP2-F12 by 25%. Furthermore, the RMSD of ωB97M-V is more than 5 times smaller than that of PBE, and nearly 4 times smaller than that of the latest hybrid Minnesota functional, MN15. These results encourage the use of ωB97M-V as a low-cost alternative for generating reference values for fitting empirical water potentials.

Our recent Topical Review of DFT, titled "Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals" has been read more then 3,000 times and is the 3rd most read article on the Molecular Physics website. It has also been tweeted 49 times according to Altmetric.
Our recent Topical Review of DFT, titled "Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals" has been read more then 4,000 times and is the 2nd most read article on the Molecular Physics website.
